Reaction: PTEN cancer mutants do not dephosphorylate PIP3
- in pathway: PTEN Loss of Function in Cancer
One of the functions of PTEN is to act as a phosphoinositide phosphatase that catalyzes dephosphorylation of PIP3 into PIP2. PTEN thus reduces the amount of available PIP3, counteracting PI3K activity and downregulating AKT signaling. PTEN is frequently targeted by loss of function mutations in cancer and in familial cancer syndromes known as PTHS (PTEN hamartoma tumor syndromes, a collection of diseases including Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, and Lhermitte-Duclos disease). Some PTEN loss-of-function variants are also found in autism spectrum disorder patients. For a recent review of PTEN involvement in cancer, please refer to Hollander et al. 2011.
The majority of missense mutations that impair phosphoinositide phosphatase activity of PTEN cluster in exon 5 of PTEN gene and result in substitution of amino acid residues in the catalytic cleft of the phosphatase domain. Arginine at position 130 is the most frequently substituted PTEN residue in cancer. R130 of human PTEN is the last arginine residue in the conserved H-C-K/R-A-G-K-G-R sequence (corresponding to HCXXGXXR motif of protein tyrosine phosphatases) in the catalytic cleft of the PTEN phosphatase domain and is essential for catalysis (Barford et al. 1994, Lee et al. 1999). PTEN R130 substitution mutants show markedly decreased phosphoinositide phosphatase activity (Han et al. 2000, Koul et al. 2002) and are frequently found in endometrial carcinoma (Kong et al. 1997, Konopka et al. 2007). The cysteine residue at position 124 (C124) of human PTEN, in the conserved H-C-K/R-A-G-K-G-R sequence, 'attacks' the phosphate group of a substrate and forms a thio-phosphate intermediate during the dephosphorylation reaction (Guan and Dixon 1991, Barford et al. 1994, Lee et al. 1999). Therefore, substitution of this critical C124 abolishes PTEN phosphatase activity (Han et al. 2000, Koul et al. 2002). Substitution of histidine H123 in the conserved H-C-K/R-A-G-K-G-R sequence also impairs PTEN phosphatase activity (Lee et al. 1999).
Missense mutations also target amino acid residues in the N-terminal phosphatase domain that are outside the catalytic cleft. Substitution of histidine at position 93 affects the conserved WPD loop of the phosphatase domain of PTEN, and PTEN H93 mutants show low phosphoinositide phosphatase activity (Lee et al. 1999). Serine residue S170 and histidine residue H173 participate in the formation of hydrogen bonds between the N-terminal phosphatase domain of PTEN and the C-terminal membrane-binding C2 domain (Lee et al. 1999). H173, and to a lesser extent S170, are targeted by missense mutations in cancer, and substitution mutants have impaired phosphoinositide phosphatase activity (Han et al. 2000).
Missense mutations also occur in the C2 domain of PTEN. The C2 domain is implicated in membrane binding and localization of PTEN, but also in PTEN roles unrelated to its phosphoinositide phosphatase function (Raftopoulou et al. 2004). Since the roles of these C2 domain PTEN mutants in cancer have not been clarified, these variants will be annotated when more information becomes available.
Besides missense mutations, nonsense mutations that result in PTEN protein truncation are also frequently found in cancer samples. The three residues most frequently targeted by nonsense mutations are R130, R233 and R335. While R130* mutation directly affects the phosphatase domain of PTEN, R233* and R335* affect the C2 domain. PTEN 130*, PTEN R233* and PTEN R335* mutants have not been functionally studied, but it was shown that comparable PTEN truncation mutants generated by directed mutagenesis, PTEN-254 and PTEN-342, were unstable when expressed in human cells and had severely diminished phosphatase activity in vitro (Georgescu et al. 1999).
Cancer-derived PTEN truncation mutants whose phosphatase domain (amino acid residues 14-185) is absent or partially truncated are annotated as truncation mutant set members (PTEN E7*, PTEN R11*, PTEN K13*, PTEN Q17*, PTEN E18*, PTEN G20*, PTEN L23*, PTEN Y27*, PTEN E40*, PTEN E43*, PTEN Y46*, PTEN S59*, PTEN K62*, PTEN Y65*, PTEN Y68*, PTEN E73*, PTEN R84*, PTEN Y88*, PTEN E91*, PTEN Q97*, PTEN E99*, PTEN C105*, PTEN Q110*, PTEN W111*, PTEN E114*, PTEN K125*, PTEN G127*, PTEN K128*, PTEN G129*, PTEN R130*, PTEN Y138*, PTEN L139*, PTEN L146*, PTEN K147*, PTEN Q149*, PTEN E150*, PTEN E157*, PTEN K163*, PTEN G165*, PTEN Q171*, PTEN Y174*, PTEN Y176*, PTEN Y177*, PTEN Y178*, PTEN Y180*, PTEN L182*).
Cancer-derived PTEN truncation mutants whose phosphatase domain is intact but whose C2 tensin-type domain (amino acids 190-350), responsible for interaction with membrane phospholipids and for the overall protein conformation (Lee et al. 1999), is missing or partially truncated are annotated as truncation mutant set candidates (PTEN R189*, PTEN G209*, PTEN C211*, PTEN Q214*, PTEN C218*, PTEN Q219*, PTEN K221*, PTEN Y225*, PTEN S229*, PTEN G230*, PTEN R233*, PTEN E235*, PTEN Y240*, PTEN E242*, PTEN Q245*, PTEN L247*, PTEN C250*, PTEN E256*, PTEN Q261*, PTEN K263*, PTEN W274*, PTEN E284*, PTEN S287*, PTEN E288*, PTEN E291*, PTEN G293*, PTEN C296*, PTEN Q298*, PTEN E299*, PTEN E307*, PTEN E314*, PTEN L320*, PTEN K330*, PTEN K332*, PTEN R335*, PTEN Y336*, PTEN K344*, PTEN Y346*).
In cancer, PTEN is also frequently inactivated by genomic deletions and loss of heterozygosity (LOH) affecting chromosome band 10q23 or by epigenetic silencing (reviewed by Hollander et al. 2011).
The majority of missense mutations that impair phosphoinositide phosphatase activity of PTEN cluster in exon 5 of PTEN gene and result in substitution of amino acid residues in the catalytic cleft of the phosphatase domain. Arginine at position 130 is the most frequently substituted PTEN residue in cancer. R130 of human PTEN is the last arginine residue in the conserved H-C-K/R-A-G-K-G-R sequence (corresponding to HCXXGXXR motif of protein tyrosine phosphatases) in the catalytic cleft of the PTEN phosphatase domain and is essential for catalysis (Barford et al. 1994, Lee et al. 1999). PTEN R130 substitution mutants show markedly decreased phosphoinositide phosphatase activity (Han et al. 2000, Koul et al. 2002) and are frequently found in endometrial carcinoma (Kong et al. 1997, Konopka et al. 2007). The cysteine residue at position 124 (C124) of human PTEN, in the conserved H-C-K/R-A-G-K-G-R sequence, 'attacks' the phosphate group of a substrate and forms a thio-phosphate intermediate during the dephosphorylation reaction (Guan and Dixon 1991, Barford et al. 1994, Lee et al. 1999). Therefore, substitution of this critical C124 abolishes PTEN phosphatase activity (Han et al. 2000, Koul et al. 2002). Substitution of histidine H123 in the conserved H-C-K/R-A-G-K-G-R sequence also impairs PTEN phosphatase activity (Lee et al. 1999).
Missense mutations also target amino acid residues in the N-terminal phosphatase domain that are outside the catalytic cleft. Substitution of histidine at position 93 affects the conserved WPD loop of the phosphatase domain of PTEN, and PTEN H93 mutants show low phosphoinositide phosphatase activity (Lee et al. 1999). Serine residue S170 and histidine residue H173 participate in the formation of hydrogen bonds between the N-terminal phosphatase domain of PTEN and the C-terminal membrane-binding C2 domain (Lee et al. 1999). H173, and to a lesser extent S170, are targeted by missense mutations in cancer, and substitution mutants have impaired phosphoinositide phosphatase activity (Han et al. 2000).
Missense mutations also occur in the C2 domain of PTEN. The C2 domain is implicated in membrane binding and localization of PTEN, but also in PTEN roles unrelated to its phosphoinositide phosphatase function (Raftopoulou et al. 2004). Since the roles of these C2 domain PTEN mutants in cancer have not been clarified, these variants will be annotated when more information becomes available.
Besides missense mutations, nonsense mutations that result in PTEN protein truncation are also frequently found in cancer samples. The three residues most frequently targeted by nonsense mutations are R130, R233 and R335. While R130* mutation directly affects the phosphatase domain of PTEN, R233* and R335* affect the C2 domain. PTEN 130*, PTEN R233* and PTEN R335* mutants have not been functionally studied, but it was shown that comparable PTEN truncation mutants generated by directed mutagenesis, PTEN-254 and PTEN-342, were unstable when expressed in human cells and had severely diminished phosphatase activity in vitro (Georgescu et al. 1999).
Cancer-derived PTEN truncation mutants whose phosphatase domain (amino acid residues 14-185) is absent or partially truncated are annotated as truncation mutant set members (PTEN E7*, PTEN R11*, PTEN K13*, PTEN Q17*, PTEN E18*, PTEN G20*, PTEN L23*, PTEN Y27*, PTEN E40*, PTEN E43*, PTEN Y46*, PTEN S59*, PTEN K62*, PTEN Y65*, PTEN Y68*, PTEN E73*, PTEN R84*, PTEN Y88*, PTEN E91*, PTEN Q97*, PTEN E99*, PTEN C105*, PTEN Q110*, PTEN W111*, PTEN E114*, PTEN K125*, PTEN G127*, PTEN K128*, PTEN G129*, PTEN R130*, PTEN Y138*, PTEN L139*, PTEN L146*, PTEN K147*, PTEN Q149*, PTEN E150*, PTEN E157*, PTEN K163*, PTEN G165*, PTEN Q171*, PTEN Y174*, PTEN Y176*, PTEN Y177*, PTEN Y178*, PTEN Y180*, PTEN L182*).
Cancer-derived PTEN truncation mutants whose phosphatase domain is intact but whose C2 tensin-type domain (amino acids 190-350), responsible for interaction with membrane phospholipids and for the overall protein conformation (Lee et al. 1999), is missing or partially truncated are annotated as truncation mutant set candidates (PTEN R189*, PTEN G209*, PTEN C211*, PTEN Q214*, PTEN C218*, PTEN Q219*, PTEN K221*, PTEN Y225*, PTEN S229*, PTEN G230*, PTEN R233*, PTEN E235*, PTEN Y240*, PTEN E242*, PTEN Q245*, PTEN L247*, PTEN C250*, PTEN E256*, PTEN Q261*, PTEN K263*, PTEN W274*, PTEN E284*, PTEN S287*, PTEN E288*, PTEN E291*, PTEN G293*, PTEN C296*, PTEN Q298*, PTEN E299*, PTEN E307*, PTEN E314*, PTEN L320*, PTEN K330*, PTEN K332*, PTEN R335*, PTEN Y336*, PTEN K344*, PTEN Y346*).
In cancer, PTEN is also frequently inactivated by genomic deletions and loss of heterozygosity (LOH) affecting chromosome band 10q23 or by epigenetic silencing (reviewed by Hollander et al. 2011).
Reaction - small molecule participants:
H2O [cytosol]
PI(3,4,5)P3 [plasma membrane]
Reactome.org reaction link: R-HSA-2317387
======
Reaction input - small molecules:
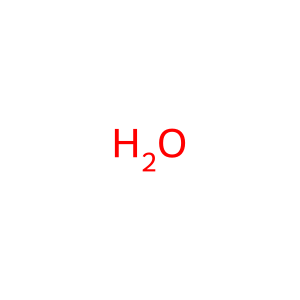
water
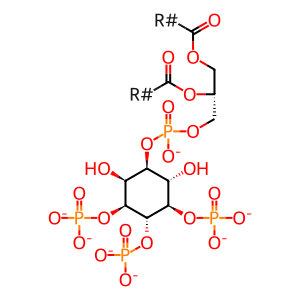
1-phosphatidyl-1D-myo-inositol 3,4,5-trisphosphate(7-)
Reaction output - small molecules:
Reactome.org link: R-HSA-2317387