Reaction: Ivacaftor:CFTR G551D transports Cl- from cytosol to extracellular region
- in pathway: Defective CFTR causes cystic fibrosis
Cystic fibrosis transmembrane conductance regulator (CFTR) is a low conductance chloride-selective channel that mediates the transport of chloride ions in human airway epithelial cells which plays a key role in maintaining homoeostasis of epithelial secretions in the lungs. Defects in CFTR can cause cystic fibrosis (CF; MIM:602421), a common generalised disorder in Caucasians affecting the exocrine glands. CF results in an ionic imbalance that impairs clearance of secretions, not only in the lung, but also in the pancreas, gastrointestinal tract and liver. Wide-ranging manifestations of the disease include chronic lung disease, exocrine pancreatic insufficiency, blockage of the terminal ileum, male infertility and salty sweat.
The class 3 mutations of CFTR such as G551D strongly decrease the time spent by CFTR in the open state (a gating defect). Results from 2-phase clinical trials using VX-770 (aka Ivacaftor), a CFTR potentiator, showed an increased CFTR channel open probability in G551D patients. Ivacaftor use showed improvements in CFTR and lung function of patients with at least one G551D allele (Accurso et al. 2010, Ramsey et al. 2011, Kapoor et al. 2014). In 2012, the FDA approved Ivacaftor (under the trade name Kalydeco) for use in cystic fibrosis patients with the G551D mutation (Ledford 2012).
The class 3 mutations of CFTR such as G551D strongly decrease the time spent by CFTR in the open state (a gating defect). Results from 2-phase clinical trials using VX-770 (aka Ivacaftor), a CFTR potentiator, showed an increased CFTR channel open probability in G551D patients. Ivacaftor use showed improvements in CFTR and lung function of patients with at least one G551D allele (Accurso et al. 2010, Ramsey et al. 2011, Kapoor et al. 2014). In 2012, the FDA approved Ivacaftor (under the trade name Kalydeco) for use in cystic fibrosis patients with the G551D mutation (Ledford 2012).
Reaction - small molecule participants:
Cl- [extracellular region]
Pi [cytosol]
ADP [cytosol]
H2O [cytosol]
ATP [cytosol]
Cl- [cytosol]
Reactome.org reaction link: R-HSA-5678992
======
Reaction input - small molecules:
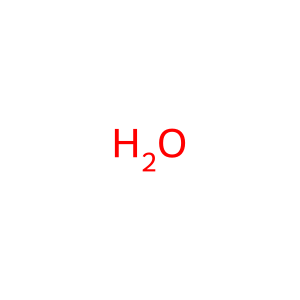
water
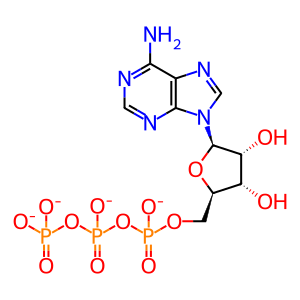
ATP(4-)

chloride
Reaction output - small molecules:
chloride
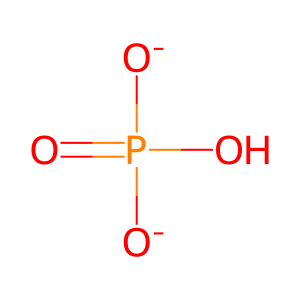
hydrogenphosphate
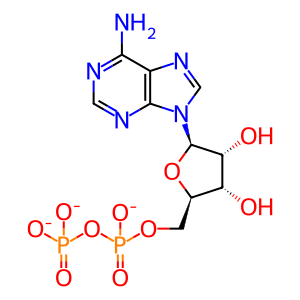
ADP(3-)
Reactome.org link: R-HSA-5678992