Reaction: Defective UGT1A1 does not transfer GlcA from UDP-GlcA to BMG
- in pathway: Defective UGT1A1 causes hyperbilirubinemia
Bilirubin (BIL) is a breakdown product of heme, causing death if allowed to accumulate in the blood. It is highly lipophilic and thus requires conjugation to become more water soluble to aid excretion. UGT1A1 is the only enzyme that converts bilirubin to either its monoglucuronide (BMG) or diglucuronide (BDG) conjugate. Defects in UGT1A1 can cause hyperbilirubinemia syndromes ranging from mild forms such as Gilbert syndrome (GILBS; MIM:143500) and transient familial neonatal hyperbilirubinemia (HBLRTFN; MIM:237900) to more severe Crigler-Najjar syndromes 1 and 2 (CN1, CN2; MIM:218800 and MIM:606785). Mutations causing CN1, the most severe form of hyperbilirubinemia, include Y293Mfs*69, S376F, R341*, G309E and Q357R (Ritter et al. 1992, Bosma et al. 1992, Maruo et al. 2003, Erps et al. 1994, Francoual et al. 2002).
Reaction - small molecule participants:
BMG [endoplasmic reticulum lumen]
UDP-GlcA [endoplasmic reticulum lumen]
Reactome.org reaction link: R-HSA-9036102
======
Reaction input - small molecules:
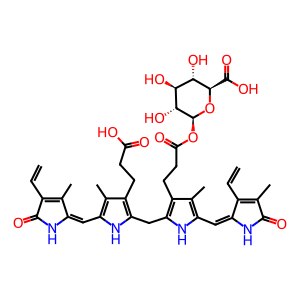
mono(glucosyluronic acid)bilirubin
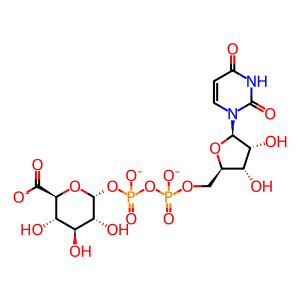
UDP-alpha-D-glucuronate(3-)
Reaction output - small molecules:
Reactome.org link: R-HSA-9036102