Reaction: Defective GAA does not hydrolyze lysosomal glycogen
- in pathway: Glycogen storage disease type II (GAA)
Normally, lysosomal alpha-glucosidase (GAA) catalyzes the hydrolysis of alpha(1,4) and alpha(1,6) linkages in glycogen, yielding free glucose and shortened glycogen polymers. A wide variety of GAA mutations reduce or eliminate this catalytic activity, leading to glycogen accumulation in lysosomes. The two missense mutant alleles annotated here encode GAA variants with little or no activity and are associated with the infantile form of the disease (early onset with multiple tissues involved) (Brown et al. 1970; Hermans et al. 1991; Zhong et al. 1991). The defect primarily affects skeletal and cardiac muscle, so the disease event is annotated here as a failure of processing of the muscle (GYG1) form of glycogen.
Reaction - small molecule participants:
H2O [lysosomal lumen]
Reactome.org reaction link: R-HSA-9036729
======
Reaction input - small molecules:
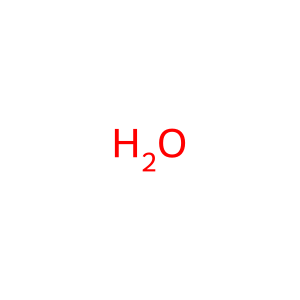
water
Reaction output - small molecules:
Reactome.org link: R-HSA-9036729